Comandos
de RasMol

Comandos
de RasMol
RasMol permite ejecutar comandos interactivos tecleados tras el inductor "RasMol" en la ventana de la línea de comandos. Las órdenes se dan siempre en líneas separadas. Se pueden utilizar tanto letras mayúsculas como minúsculas para los comandos, ya que es insensible a esta característica. Los espacios en blanco son ignorados excepto en aquellos casos en los que sirvan para separar las ordenes de sus argumentos.
A continuación presentamos una lista
de los comandos y claves reconocidos actualmente por RasMol.
Para una información más detallada
sobre cada una de las funciones de RasMol teclee "help <command".
Sintaxis: backbone {<boolean}
backbone <valor
backbone dashes
La orden "backbone" de RasMol hace posible la representación del esqueleto del polipéptido, como una serie de enlaces, representados como cilindros, que conectan los carbonos alfas adyacentes de cada aminoácido. En el caso de ácidos nucleicos, el comando "backbone" activa la representación del esqueleto de la molécula, como una serie de enlaces, en forma de cilindros, que conectan los grupos ribosa o desoxiribosa fosfato adyacentes de cada nucleótido.
La visualización de estos enlaces a
lo largo del eje de la molécula se activa o desactiva con el parámetro
de la orden; con la orden "backbone off" se desactivan los "enlaces" seleccionados
y con "backbone on" o "backbone (valor)" se activan.
|
 |
Este mismo efecto puede lograrse mediante el Menú de Herramientas en la Ventana Display.
El número puede ser utilizado para especificar
en unidades angstróms o unidades de rasmol el radio del cilindro
de la representación. Un valor de parámetro de 500 (2.0 angstróms)
o mayor puede resultar en un error que aparecerá como "Valor del
parámetro demasiado grande" (Parameter value too large). Los elementos
de la representación se pueden colorear utilizando el comando de
RasMol "colour backbone".
Sintaxis: background <colour
La orden de RasMol "background" selecciona
el color de fondo en la ventana RasMol. El color puede ser determinado
a través del nombre del color o por medio de componentes triples
de Rojo, Verde y Azul (RGB) separados por comas y delimitados por corchetes.
Al teclear la orden "help colours" se obtendrá una lista de los
nombres de colores predefinidos y reconocidos por RasMol. Si se utiliza
X Windows, RasMol es capaz de reconocer aquellos colores que se encuentran
en la base de datos de nombres de colores del servidor X.
Sintaxis: cartoon <number
La orden de RasMol "cartoon" permite la representación
de la molécula en forma de cintas para permitir mostrar la representación
de Richardson (MolScript). Actualmente se implementan como cintas delgadas.
La forma más simple de obtener este tipo de representación
es su uso desde el Menú de Herramientas en la ventana "Display".
|
 |
Cuando la orden es utilizada desde la línea
de comandos, la representación se observa sobre aquellas regiones
preseleccionadas de la molécula. Si no se específica ningún
parámetro o valor luego del comando "cartoons", el ancho de las
cintas tomará del tipo de estructura secundaria de la proteína.
Por defecto, el extremo C-terminal de las hojas beta se representará
como la cabeza de la flecha.
Sintaxis: center {<expression}
centre {<expression}
La orden "center" determina el punto alrededor
del cual puede gira la molécula, bien sea mediante el comando "rotate"
o mediante las barras de desplazamiento en la Ventana de Gráficos
Principal de RasMol. Cuando no se específica ningún parámetro,
el comando "centre" toma el centro de gravedad de la molécula
o de los átomos preseleccionados como eje de rotación.
|
 |
Si la expresión especifica un único
átomo como eje de rotación, este átomo permanecerá
"inmóvil" durante las rotaciones que se realicen sobre la molécula
en cuestión.
Sintaxis: clipboard
La orden de RasMol "clipboard" coloca la imagen
representada en un determinado momento en "clipboard" de gráficos
local. Esta orden aún no se puede utilizar con los sistemas UNIX
o VMS; está ideada para hacer más fácil la transferencia
de imágenes entre aplicaciones bajo Microsoft Windows o Apple Macintosh.
Si se está operando con el programa RasMol sobre los sistemas UNIX
y VMS, esta función se puede realizar generando una "raster image"
en un formato que el programa receptor puede leer usando la orden de RasMol
"write".
Sintaxis: colour {<object} <colour
color {<object} <colour
color {<object} <[RGB triplet]
Esta orden sirve para colorear los átomos (u otros elementos) de la región seleccionada. El color puede ser determinado a través del nombre del color o de componentes triples de Rojo, Verde y Azul (RGB) separados por comas y delimitados por corchetes. Un triplete típico es [255,255,255] que representa el color blanco. Al teclear el comando "help colours" se obtendrá una lista de los nombres de colores predefinidos reconocidos por RasMol.
Los objetos permitidos para colorear son "atoms", "bonds", "backbone", "ribbons", "labels", "dots", "hbonds", y "ssbonds", si no se específica ningún objeto, por omisión, se adopta "átom".
|
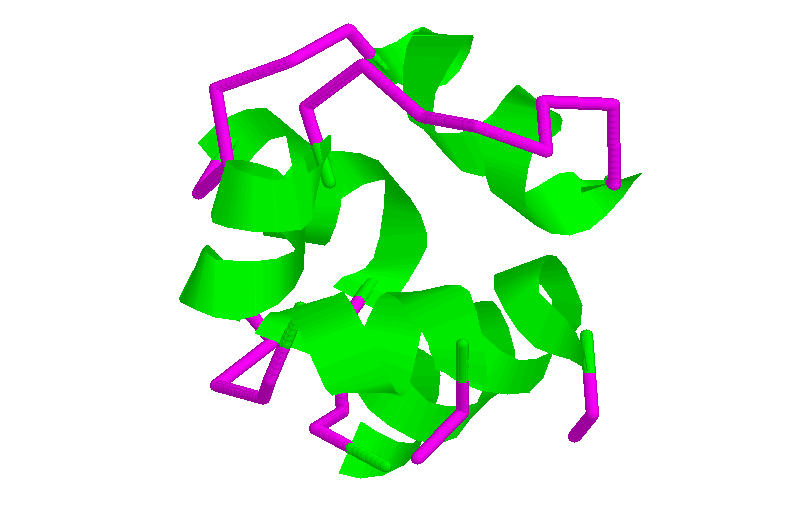 |
El comando "color" permite diferentes esquemas definidos como "cpk", "amino", "chain", "group", "shapely", "structure", "temperature", "charge", y "user". Este mismo efecto puede lograrse mediante el Menú de Herramientas en la Ventana Color.
Consulte el Manual de Rasmol : Sección
Colores y Esquemas de Color
Sintaxis: connect {<boolean}
La orden de RasMol "connect" obliga a RasMol
a recalcular la conectividad de la molécula con la que estamos trabajando.
Si el archivo original de entrada de datos contenía información
sobre la conectividad, se descarta. La orden "connect false" utiliza un
algoritmo heurístico muy rápido adecuado para determinar
el enlace en grandes biomoléculas tales como proteínas o
ácidos nucléicos. La orden "connect true" usa un algoritmo
más lento pero más exacto basado en radios covalentes que
es más apropiado para moléculas que contienen elementos inorgánicos
o "anillos tensionados". Si no se introduce ningún parámetro,
RasMol determina qué algoritmo utilizar tomando como base el número
de átomos en el archivo. Una cantidad mayor que 255 átomos
hace que RasMol utilice la ejecución más rápida. Este
es el método aplicado para determinar el enlace, si es necesario,
cuando una molécula es leída en primer lugar utilizando la
orden "load".
Sintaxis: define <identifier <expression
La orden "define" permite al usuario asociar
un grupo arbitrario de átomos con un identificador único.
Esto hace posible la construcción de grupos definidos por el usuario.
A estos grupos se les declara estáticamente, es decir, una vez definidos,
el contenido de los grupos no cambian, incluso si la expresión que
los define depende de las transformaciones llevadas a cabo en ese preciso
momento o sí se modica la representación de la molécula.
Sintaxis: dots {<boolean}
dots <value
La orden "dots" se usa con el fin de generar una superficie de puntos Van der Waal alrededor de los átomos seleccionados en un determinado momento. Las superficies de puntos visualizan puntos a una distancia regular en una esfera de radio Van der Waals alrededor de cada átomo seleccionado. Aquellos puntos que estarían "enterrados" dentro del radio de Van der Waals de cualquier átomo (seleccionados o no) no se visualizan. La orden "dots on" borra cualquier superficie de puntos existentes y genera una superficie de puntos alrededor del conjunto de átomos actualmente seleccionados con una densidad de error de puntos de 100. El comando "dots off" borra cualquier superficie de puntos existente. Podemos especificar la densidad de puntos proporcionando un parámetro numérico entre 1 y 1000. Este valor corresponde aproximadamente al número de puntos que aparece en la superficie de un átomo de tamaño medio.
Por defecto, el color de cada punto en la superficie
de puntos es el mismo que el del átomo que se encuentre más
próximo a él en el momento de generar la superficie. Con
la orden "colour dots" podemos cambiar el color de la superficie de puntos
completa.
Sintaxis: echo {<string}
La orden de RasMol "echo" se utiliza para visualizar
un mensaje en la pantalla del terminal de RasMol. El parámetro de
cadena puede ser opcionalmente delimitado en caracteres separados por comillas.
Si no especificamos ningún parámetro, la orden echo visualiza
una línea en blanco. Este comando es especialmente útil a
la hora de visualizar un texto del archivo de un script de RasMol.
Sintaxis: exit
El comando "exit" de RasMol se usa para terminar
la ejecución de un script (volviendo a la línea de órdenes,
o el script que lo había llamado).
Sintaxis: hbonds {<boolean}
hbonds <value
La orden de RasMol "hbonds" se utiliza para representar los enlaces de hidrógeno del eje de la molécula. Esta información es particularmente útil para visualizar la estructura secundaría en proteínas. Los enlaces de hidrógenos se representan como líneas punteadas o cilindros entre los residuos donante y aceptante. La primera vez que se usa la orden "hbonds", el programa se encarga de buscar la estructura de la molécula con el fin de encontrar resíduos enlazados de hidrógenos y después de informar al usuario del número de enlaces. La orden "hbonds on" visualiza los enlaces seleccionados como líneas punteadas y el comando "hbonds off" los desactiva.
|
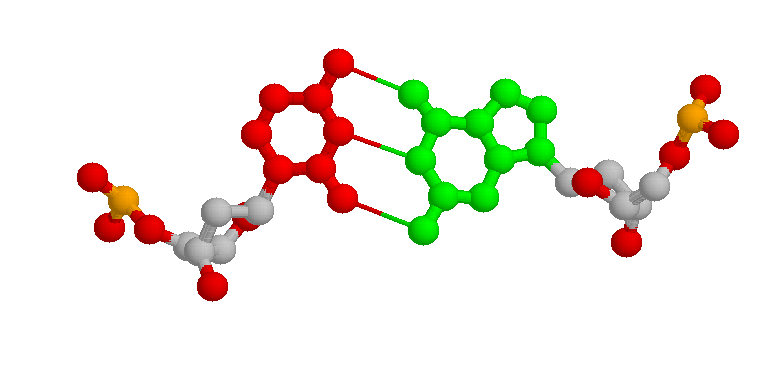 |
Para cambiar el color de los elementos del
enlace hidrógeno se emplea el comando "colour hbonds". En principio,
cada enlace hidrógeno es del color de los átomos conectados
con él.
Sintaxis: help {<topic {<subtopic}}
? {<topic {<subtopic}}
La orden de RasMol "help" provee ayuda en línea
(on-line) sobre un tema concreto.
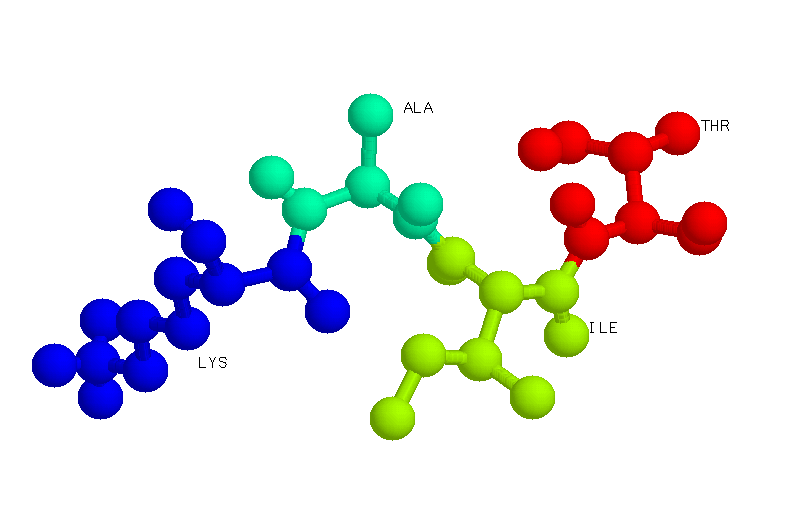
Sintaxis: label {<string}
label <boolean
El comando "Label" permite asignar y visualizar una línea de texto sobre uno o varios átomos seleccionados en la molécula de interés. Este comando contiene especificadores de expansión, que visualizan las propiedades del átomo al que se está etiquetando. Un especificador de expansión consiste en un carácter "%" seguido de un único carácter alfabético que especifica la propiedad que va a ser visualizada.
Los especificadores que pueden acompañar
a comando "label" son
|
|
Nombre del átomo. |
|
|
Factor B/temperatura. |
|
|
Identificador de cadena. |
|
|
Símbolo del elemento atómico. |
|
|
Número serial del átomo. |
|
|
Código de una letra del aminoácido. |
|
|
Código de tres letras del aminoácido. |
|
|
Número de resíduo. |
|
 |
Los textos generados en la ventana del Rasmol
pueden desactivarse con el comando "label off". La orden "colour label"
hace posible el cambio de color del texto. Por defecto, el texto aparece
con el mismo color que el del átomo al que está adjunto.
Es posible cambiar el tamaño del texto visualizado con la orden
"set fontsize".
Sintaxis: load {<format} <nombre
de archivo
load {<format} inline
Este comando carga un archivo de coordenadas moleculares en RasMol. Los formatos válidos son Brookhaven Protein Databank (PDB), Alchemy de Tripos Associates y Sybyl Mol2, Mol de Molecular Design Limited (MDL), Minnesota Supercomputer Centre's (MSC) XYZ (XMol) y CHARMm. También mopac (formato de archivo mopac; bien sea cartesiano o formato de z-matrix), nmrpdb (formato de archivo multi-pdb nmr). Si no se específica el formato, se asume que por defecto será pdb. Solo una molécula puede ser visualizada en un momento dado. Antes de cargar una nueva molécula hay que eliminar la anterior con la orden de RasMol "zap".
La orden "load" selecciona todos los átomos
de la molécula, centrándola en la pantalla y produciendo
un modelo coloreado (colores CPK) en el modelo de cilindros (wireframe).
Sintaxis: monitor <number <number
monitor {<boolean}
La orden "monitor" de RasMol permite activar la visualización de monitor de distancia. Un monitor de distancia es una línea discontinua entre un par cualquiera de átomos, opcionalmente etiquetado con la distancia que separa a ambos. La orden "monitor <número <número" añade tal monitor de distancia entre los dos átomos indicados por los números dados como parámetros.
Los monitores de distancia se desconectan con el comando *monitors off*. Por defecto, los monitores visualizan la distancia entre sus dos puntos extremos como una etiqueta en el centro del monitor. Estas etiquetas de distancia pueden ser desconectadas mediante la orden *set monitors off* y reactivados con *set monitors on*. Como muchas de las demás manifestaciones el color de un monitor se toma del color de sus extremos a menos que se especifique con la orden *colour monitors*.
Los monitores de distancia pueden ser añadidos
a una molécula interactivamente, con el ratón, usando el
comando *set picking monitor*. Haciendo click sobre un átomo produce
su identificación en la línea de comandos. Además
cada átomo picado produce un incremento de un módulo contador
tal como el modo monitor y cada segundo átomo visualiza la distancia
entre este y el picado previamente. La tecla de desplazamiento puede usarse
para formar monitores de distancia entre el átomo fijado y distintas
posiciones consecutivas. Un monitor de distancia puede también eliminarse
seleccionando el par apropiado de átomos (los unidos en el monitor
de distancia) una segunda vez.
Sintaxis: pause
La orden "pause" de RasMol se usa para la edición
de Scripts, este comando detiene momentáneamente la ejecución
de las ordenes siguientes hasta que no se oprima cualquier tecla. Durante
la pausa, RasMol permite la manipulación de la imagen usando el
ratón y las barras de desplazamiento, también permite ajustar
el tamaño de la Ventana de RasMol y de la Línea de Comandos.
Sintaxis: print
La orden de RasMol "print" envía la
imagen actualmente visualizada a la impresora local, si no se especifica
otra, utilizando el controlador original de la impresora del sistema operativo.
Este comando todavía no se puede utilizar con los sistemas UNIX
o VMS. Esta ideado para la utilización y aprovechamiento de los
controladores de impresora de Microsoft Windows y Apple Macintosh. Así,
por ejemplo, hace posible imprimir una imagen directamente en una impresora
de matriz de puntos. Si utilizamos RasMol con los sistemas UNIX y VMS esta
función se puede llevar a cabo creando un archivo PostScript con
los comandos write ps o write vectps e imprimiéndolo posteriormente;
o bien mediante la creación de un archivo de imágenes exportables
y utilizando alguna herramienta para mandarlo a la impresora local.
Sintaxis: quit
Salir del programa RasMol. El comando de RasMol
"exit" tiene una función diferente.
Sintaxis: refresh
La orden de RasMol "refresh" se usa en archivos
script para redibujar la imagen local. Este comando se utiliza principalmente
en la creación de scripts de RasMol.
Sintaxis: reset
La orden de RasMol "reset" restaura la transformación
de la imagen original y el centro de la rotación. La escala se sitúa
en su valor por defecto, zoom 100; el centro de rotación se sitúa
en el centro geométrico de la molécula actualmente cargada.
Es importante no confundir éste con la orden de RasMol "zap", que
borra la molécula almacenada actualmente, volviendo al estadio inicial
del programa.
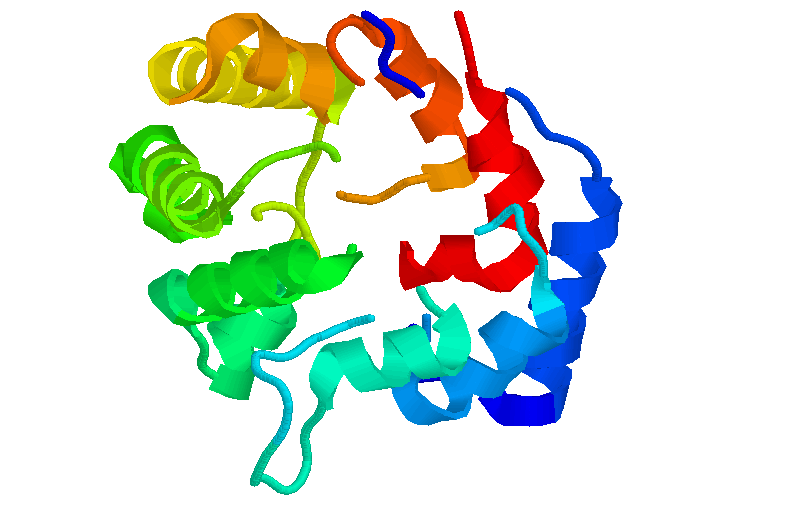
Sintaxis: restrict {<expression}
La orden de RasMol "restrict", permite definir y limitar una región de interés dentro de la molécula e igualmente desactivar cualquier tipo de representación en el resto de la molécula. Los comandos posteriores que se ejecutan desde la Línea de Comandos o desde la Barra de Herramientas en la ventana RasMol sólo modificarán la región definida inicialmente.
|
 |
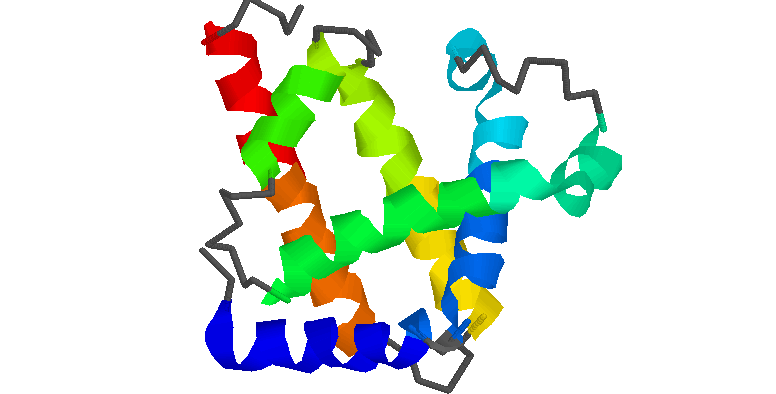
Sintaxis: ribbons {<boolean}
ribbons <value
La orden de RasMol "ribbons" visualiza la proteína o ácido nucléico actualmente cargados como una superficie de "cintas" densa y lisa que pasa a lo largo del eje de la molécula, entre los carbonos alfa de los aminoácidos si se trata de una proteína ó entre los grupos pentosa-fosfato de los nucleótidos seleccionados si se trata de una ácido nucléico.
Podemos cambiar el color de la cinta con la orden de RasMol "colour ribbon". Si el color es none (ninguno), el preestablecido, por defecto la cinta toma el color de átomo que se encuentre a su misma altura.
El ancho de la cinta está determinado por la estructura secundaría definida en el archivo PDB. Para proteínas el ancho de las alfas hélices y de las hojas plegadas beta es de 380 (1,52 angstróms) y de 100 (0,4 angstróms) respectivamente y para los giros y las hélices al azar es de 100 (0,4 angstróms). Para los ácidos nucléicos, el ancho de la cinta tiene un valor constante de 720 (2,88 angstróms).
|
 |
Sintaxis: rotate <axis {-} <value
Esta orden hace girar la molécula alrededor
del eje especificado. Los valores permitidos para el eje son "x", "y" y
"z". El parámetro entero establece en grados el ángulo que
la estructura rotará. En el caso de los ejes X y Y, los valores
positivos desplazan el punto más cercano hacia la derecha y hacia
arriba, mientras que los valores negativos lo desplazan hacia la izquierda
y hacia abajo. En cuanto al eje Z, una rotación positiva actúa
en la dirección de las agujas del reloj y una negativa en sentido
opuesto.
Sintaxis: save {pdb} <nombre de archivo
save alchemy <nombre de archivo
save mdl <nombre de archivo
Guarda el grupo de átomos seleccionados
actualmente en un fichero Brookhaven Protein Database (PDB) o un archivo
de formato Alchemy (tm). Si no se específica la extensión
del archivo el comando "save" crea un archivo "PDB", mientras que el comando
"write" genera una imagen "GIF".
Sintaxis: script <nombre de archivo
El comando de RasMol "script" lee un conjunto de órdenes RasMol secuencialmente a partir de un fichero de texto y los ejecuta. Ello permite ejecutar secuencialmente las órdenes que usamos, y efectuarlas con un único comando. Un archivo script puede contener un segundo comando llegando a una "profundidad" máxima de 10, lo que permite secuencias complicadas de acciones a ejecutar.
La forma más común de crear un archivo script de RasMol es por medio de las órdenes "write script" ó "write rasmol" con el fin de producir la secuencia de órdenes que se necesitan para volver a generar la imagen actual, la representación y los colores de la molécula visualizada en ese momento. Tales ficheros generados automáticamente producen solo una imagen.
Se pueden, también, crear archivos script de RasMol manualmente con un editor de texto. Tales archivos de script, con el uso de "pause" y "refresh", pueden producir "animaciones moleculares". La explicación de la forma de realizar y editar estos archivos "script" en el Manual de RasMol
Sección Guía para la Creación
de Scripts
Sintaxis: select {<expression}
Define una parte de la molécula sobre la cual se va a realizar alguna modificación; en lo sucesivo, todos los tipos de representación y colores asignados mediante comandos ó por la barra de herramientas en la ventana de RasMol afectarán únicamente a la región seleccionada.
Con el comando "select" es posible discriminar átomos, grupos, regiones específicas o cadenas completas dentro de una macromolécula.´La explicación detallada de todas las expresiones y definiciones de las regiones de una molécula en el Manual de RasMol
Sección
Expresiones Atómicas
Sección Grupos
Predefinidos
Sintaxis: set <parameter {<option}
El comando de RasMol "set" permite al usuario cambiar algunos parámetros internos del programa tales como los que controlan las opciones de representación. Cada parámetro posee una serie propia de opciones de parámetros permisibles. Normalmente, la omisión de la opción parámetro reajusta dicho parámetro a su valor por defecto. La descripción completa de los comandos "set" se encuentra en el Manual de RasMol
Sintaxis: show information
show sequence
show symmetry
La orden de RasMol "show" muestra detalles
del estado de la molécula con la que trabajamos. El comando "show
information" proporciona el nombre de la molécula, la clasificación,
el código PDB y el número de átomos, cadenas y grupos
que contiene. En caso de que se hayan determinado el enlace de hidrógeno,
los puentes de disulfuro o la estructura secundaria, el número de
enlaces por puente de hidrógeno, enlaces disulfuro, hélices,
plegamientos y giros beta se visualizan respectivamente. El comando "show
sequence" facilita una lista de los residuos que componen cada cadena de
la molécula.
Sintaxis: slab {<boolean}
slab <value
El comando "slab" permite realizar un corte
sobre el eje Z de la molécula, lo cual permite mostrar segmentos
y/o residuos ubicados al interior de su estructura. Es posible definir
un valor entre 0 y 100 el cual determina la posición plano de corte,
a partir de este momento solo se visualizarán las porciones de la
molécula que encuentren más alejadas que el plano de corte
respecto al observador, así el comando "slab 0" corresponde a un
corte sobre la parte posterior de la molécula y el comando "slab
100" a uno sobre su punto más delantero, valores intermedios determinan
el porcentaje de la molécula que va a ser dibujada.
Sintaxis: spacefill {<boolean}
spacefill temperature
spacefill user
spacefill <value
La orden de RasMol "spacefill" (espacio relleno)
se usa para representar todos los átomos actualmente seleccionados
como esferas sólidas. Este comando se emplea para producir tanto
esferas unidas como modelos de bolas y barras de la molécula. El
comando por defecto, representa cada átomo como una esfera con su
respectivo radio de Van der Waal. El comando "spacefill off" anula la representación
de los átomos seleccionados como esferas. Se puede especificar el
radio de la esfera como un número entero en unidades RasMol (1/250
angstróm) o un valor que contenga punto decimal. Un valor igual
o superior a 500 (2.0 angstróms) produce un error del tipo "Parameter
value too large" (valor de parámetro demasiado grande).
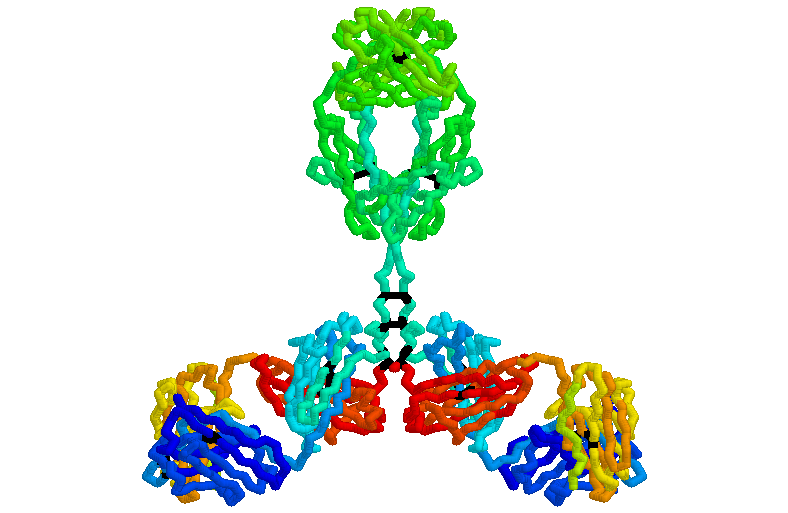
Syntax: ssbonds {<boolean}
ssbonds <value
La orden "ssbonds" se utiliza para representar los puentes disulfuro de la molécula de proteína, bien como líneas punteadas, bien como cilindros entre las cisteínas conectadas. La primera vez que se usa el comando "ssbonds", el programa busca la estructura de la proteína con el fin de encontrar pares de cisteínas (cuyos grupos sulfuros estén a unos 3 angstróms de la otra) e informa al usuario del número de puentes. La orden "ssbonds on" visualiza los enlaces escogidos como líneas de puntos y "ssbonds off" desactiva la visualización en el área seleccionada. El color de los enlaces de disulfuro se puede cambiar con la orden "colour ssbonds". Por defecto, los enlaces disulfuro se dibujan entre los átomos de sulfuro del grupo cisteína y con el mismo color de los átomos de azufre que lo forman.
|
 |
Syntax: stereo on
stereo [-] <number
stereo off
La orden "stereo" de RasMol activa una visualización de imágenes estéreo lado con lado. Esta visualización puede conectarse y desconectarse bien mediante el menú de opciones (Options menu) seleccionando Stereo, o bien tecleando la orden "stereo on" o "stereo off". El ángulo de separación entre las dos vistas puede regularse con la orden "set stereo [-] <number", donde los valores positivos producen una visión "bizca" y los negativos "relajada".
El comando "stereo" solo está parcialmente
implementado. Cuando se activa la imagen estéreo no queda bien centrada,
esto se puede arreglar con la orden "translate x -<number". Tampoco
está soportada en el vector de archivos de salida PostScript, no
es salvada por la orden "write script" y en general no esta bien intercomunicadas
con diversas otras funciones del programa. Cuando se activa slab junto
a estereo, la imagen resultante no es una visión estereoscópica.
Syntax: strands {<boolean}
strands <value
strands dashes
La orden de RasMol "strands" visualiza la proteína o ácido nucléico con que trabajamos como una "cinta" lisa curvada "depth-cued" que pasa a lo largo del eje de la molécula. La cinta está compuesta de una serie de filamentos que corren paralelos entre sí a lo largo del eje de cada aminoácido o nucleótido según sea el caso. Este mismo efecto puede lograrse con la Barra de herramientas de RasMol en la ventana "Display".
Por defecto la cinta adopta el color del átomo con el que se encuentre en su recorrido longitud. La orden "colour ribbon" cambia el color de la cinta. El filamento central y aquél que se encuentra en la parte más externa se pueden colorear independientemente con las órdenes "colour ribbon1" y "colour ribbon2", respectivamente.
El parámetro opcional determina la anchura
de la cinta en cada posición en unidades RasMol. La cinta adquiere
una anchura extraída de la estructura secundaria de la proteína
o de un valor constante de 720 para ácidos nucléicos (lo
cual significa una anchura de 2,88 angstróms). La anchura inicial
de las alfa hélices de proteína y de las hojas plegadas beta
es de 380 (1,52 angstróms) y 100 (0.4 angstróms) para giros
y espirales. Esta orden es parecida a "ribbons" que representa la cinta
como una superficie sombreada lisa.
Syntax: structure
La orden "structure" calcula las asignaciones
de estructura secundaría para la proteína cargada. En primer
lugar se localizan los enlaces de hidrógenos de la molécula,
en caso de que no se hubiera hecho previamente, la estructura secundaría
es determinada a través del algoritmo DSSP de Kabsch y Sander. Una
vez finalizado, el programa informa del número de hélices,
filamentos y giros encontrados.
Syntax: trace {<boolean}
trace <value
trace temperature
La orden de RasMol "trace" visualiza una línea
lisa entre dos carbonos alfa consecutivos. Esta línea no pasa exactamente
a través de la posición del carbono alfa de cada resíduo,
sino que sigue la misma vía de ribbons, strands, y cartoons. Cada
residuo puede ser visualizado como "ribbon", "strands", "cartoon" o "trace",
activando una de esas representaciones se inactivan las otras. Sin embargo,
un residuo puede visualizarse simultáneamente como una combinación
de "backbone" y cualquiera de las otras representaciones. El comando "trace
temperature" permite visualizar la columna como un cilindro más
ancho para factores de alta temperatura y más delgado para factores
de baja. Esta representación es útil para cristalografía
y espectroscopía de RMN.
Syntax: translate <axis {-} <value
La orden de RasMol "translate" mueve la posición
del centro de la molécula en la pantalla. El parámetro de
eje específica a lo largo de qué eje se va a trasladar la
molécula y el parámetro entero especifica la posición
absoluta del centro de la molécula desde la parte central de la
pantalla. X, Y y Z son los valores permitidos para el parámetro
eje. Los valores de desplazamiento deben oscilar entre -100 y 100, más
allá de ellos, la molécula desaparece de la pantalla. Un
desplazamiento positivo X mueve la molécula hacia la derecha; y
uno positivo Y hacia abajo. Las órdenes "translate x 0" y "translate
y 0" centran la molécula en la pantalla.
Syntax: wireframe {<boolean}
wireframe <value
wireframe dashes
La orden "wireframe" representa cada enlace de la región seleccionada como un cilindro, una línea o un vector depth-cued. Las órdenes "wireframe" o "wireframe on" activan la visualización de los enlaces a manera de vectores depth-cued (aparecen más oscuros cuanto más lejos estén del observador).
Los enlaces seleccionados se visualizan como cilindros especificando un radio como un número entero en unidades RasMol o como decimal en angstróms. Un valor de parámetro de 500 (2.0 angstróms) o más resulta en un error de "valor de parámetro demasiado grande" ("Parameter value too large"). La orden "colour bonds" colorea los enlaces.
Por defecto, cuando se carga una molécula,
por ejemplo en formato pdb, esta aparece en la ventana Rasmol en representación
tipo "wireframe". Las representaciones wireframe, backbone y strands pueden
visualizarse con líneas "borradas" (dotted). Esto se consigue con
los parámetros "dash" o "dashes" de los comandos "wireframe", "backbone"
y "strands".
Syntax: write {<format} <nombre de archivo
Este comando sirve para copiar la imagen en un archivo en un formato de exportación estándar. Los formatos de archivos de imágenes actualmente disponibles son: "gif" (Compuserve GIF), "iris" (formato IRIS RBG), "ppm" (Portable Pixmap), "ras" (Archivo exportable de Sun), "ps" y "epsf" (PostScript Encapsulado), "monops" (PostScript Monochrome Encapsulado), "vectps" (Vector PostScript, ver mas adelante), "bmp" (mapa de bits de Microsoft) y "pict" (Apple PICT). La orden "write" sirve también para crear órdenes "scripts" para otros programas de gráficos.
No existe ya ninguna diferencia entre esta
orden y la orden de RasMol "save". La única diferencia consiste
en que "save", sin un especificador de formato, genera un archivo PDB y
la orden "write" genera una imagen GIF. La orden "set write" activa y desactiva
el uso de "save" y "write" en "scripts".
Syntax: zap
Esta orden borra el contenido de la base de
datos actual y vuelve a colocar las variables de parámetros en su
estado inicial.
Syntax: zoom {<boolean}
zoom <value
Esta orden altera el aumento de la imagen visualizada.
Un valor entero entre 10 y 200 señala el aumento deseado como un
porcentaje de la escala inicial, el parámetro mínimo es 10,
el máximo depende del tamaño de la molécula que se
está visualizando, para una proteína de tamaño medio
es de aproximadamente 500.
|
|
|
|
|
|